
(Photo by Randy Montoya)
Download 300dpi JPEG image, ‘Sorin.jpg’, 1.8Mb
{kind=link}
ALBUQUERQUE, N.M. — That proteins fold isn’t just a good idea, it’s the law — the law of life.
Proteins are born resembling strings of beads. These fold to become brain, blood, biceps, and bone. But incomplete folds cause disabling diseases like Alzheimer’s and “mad cow” syndrome. They also cause respiration and locomotion failures — aims of biowarfare — because protein functions can’t be fully carried out.
Now the first successful computational model to capture the process of one protein interfering with the folding of another has been created at the Department of Energy’s Sandia National Laboratories.
The results will help laboratory scientists understand the mechanisms by which incomplete folds occur, in order to prevent them.
“This is a step toward successful protein engineering,” says principal researcher Sorin Istrail. “It provides our first clue in how to design sequences of laboratory proteins that can survive the essential but complicated folding process.”
“Our next project is to apply our system to uncover clues about the mechanism of misfolding that occurs in Alzheimer’s,” he says.
Istrail collaborated with biologist Jonathan King and computer graduate student Russell Schwartz, both from the Massachusetts Institute of Technology. A paper describing the work will be published this spring in the Journal of Computational Biology (Vol. 6, No. 2).
Says King, “This is the first time any computer scientist has taken this fundamental step within a mathematically sound model. Though there are millions of proteins in any real-world experiment, the conceptual leap in modeling is in going from one protein to two.”
King, president of the Biophysical Society, pioneered “wet” laboratory experiments to uncover protein-misfolding mechanisms.
The conceptual leap
The notorious complexity of simulating the folding of even a single protein, and the lack of computational resources to do so, had inhibited researchers from attempting robust (reproducible) multiprotein simulations till now.
Says King, “There are at least 50 research groups of biophysicists who represent a protein as a string of beads. The scientists do operations that have the [simple] protein fold back on itself. But the process fails when applied to real proteins. Why? Because if your model has only one protein, then your model doesn’t encompass reality. Two protein molecules can stick to each other instead of folding separately.
“What Sorin has done is to say, ‘If bumping is important, I will have a pair of strings move in space and bump into each other.’ He quickly discovered properties of folding that were absent in previous simulations.”
The problem was lessened by advanced algorithms and computer power available at Sandia.
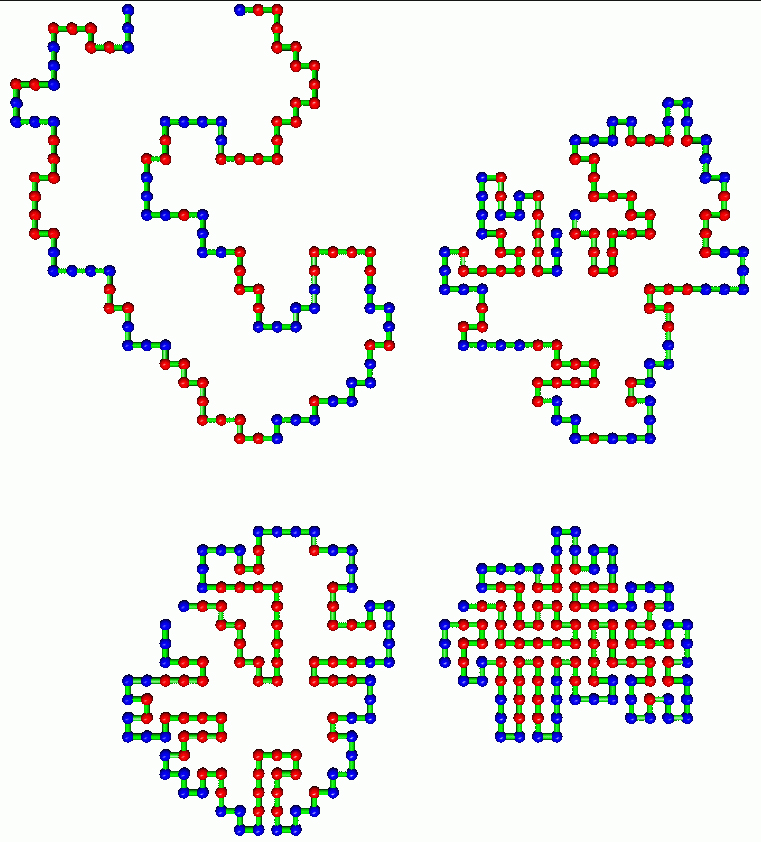
Download 150dpi JPEG image, ‘protected-pathway.jpg’, 473K
To fold well or not to fold well: that is the question
Says Istrail, “Jonathan [King], a laboratory biologist, proposed the problem. He said that computer models constructed to describe the proper folding of a single protein lack touch with experimental realities. Now his laboratory results are explained by our computational model. Russell and I designed the misfolding simulation model, and validated its robustness by varying parameters on the models.”
The researchers demonstrated that certain molecular arrangements, formerly thought random, have functions. The investigators tracked two highly simplified proteins interacting on a grid during millions of computerized trials. They found it was the apparently random positioning of water-loving (hydrophilic) molecules in a protein that prevents water-hating molecules (hydrophobic, possessors of the dominant folding force) from binding with other proteins.
“We learned that apparent randomness in the length and placement of a protein’s molecular sub-sequences is necessary to form good folds. A protein that violates this quality of apparent randomness tends to aggregate with other proteins to form inert lumps,” says Istrail, referring to the tendency of proteins that bind promiscuously to form something that resembles clumps of spaghetti.
A walk on the wild side
In Alzheimer’s and other protein-based diseases, an amino acid from one protein links to an amino acid on a second protein, rather than waiting to link to one on its own chain.
This protein “adultery,” which occurs as each protein passes through a series of intermediate folding steps, results in proteins stuck to each other in inert masses.
It is easy to see why proteins might grab at each other. Proteins are not “bad” in character but consider how they were raised: as a series of simple amino acids. Suppose, in a pile of proteins, an amino acid from one protein just happens to be lying athwart its natural partner — but that partner is a member of a different protein. Why should the first amino acid wait until the proper amino acid, pre-selected for it from its own group, comes along to tie the knot?
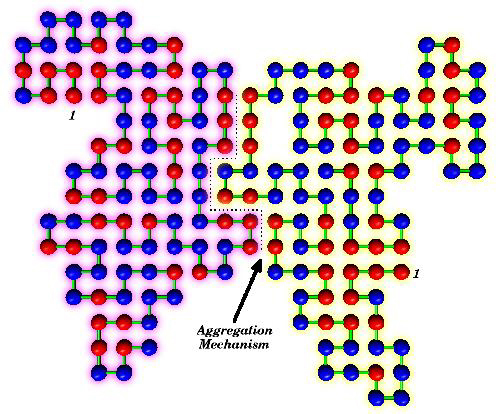
Download 72dpi JPEG image, ‘aggregation-mechanism.jpg’, 338K
Why most proteins are faithful
The researchers found that, in an aqueous solution, amino acids usually hook up with others in their own string because of the randomness with which water-hating amino acid molecules are interspersed among water-loving ones on the same protein. The water-hating molecules are prevented from combining with their counterparts on other strings by the action of water-loving molecules, which herd hydrophobics into the center of the molecule — in effect, in the last century’s terminology, circling the wagons around the women and children. The mechanism relies upon the hydrophilic protein’s love of water and the hydrophobic’s distaste for it. This draws one group of proteins forward, and pushes the other back.
Even at the start of this molecular square dance, the researchers found that the water-loving molecules form little pockets of protection around neighboring water-hating molecules. These pockets require another protein to have exactly the right shape to inject a corresponding amino acid into the tiny coastal boundaries created by the protectors. This rarely happens.
“So the apparent randomness of the distribution of water-hating and water-loving molecules on the protein chain has its reason,” says Istrail.
Think of celebrities and bodyguards, he urges. For instant protection, “You want the guardians interspersed among the celebrities, not a chain of guardians and a chain of celebrities.”
Ultimately, the “celebrities” come together to form a hydrophobic molecular core while the “guardians” line the edges of the molecule.
“Our Monte Carlo [statistical] simulations unveil how the kinetic competition between folding and aggregation is resolved by the ‘good’ sequences’ built-in protection against aggregation, due to their hydrophobic-hydrophilic pattern,” says Istrail. “This allows them to escape misfolding traps and move towards the hypothesized ‘fast track’ folding that takes place for real proteins.
“If, by repetitively folding an ideal protein, it reaches final form [native conformation] the overwhelming number of times, that’s good. If the majority of results don’t reach final form, that’s bad. This develops a sense of what characteristics are necessary for a good sequence,” says Istrail.
The Sandia-MIT simulation was based on a model created by Ken Dill of the University of California at San Francisco. This model, called HP for hydrophobic-hydrophilic, “is one of the most fundamental, and probably the most studied, biophysical lattice protein folding model anywhere,” says Istrail. “It was the vast literature on this model that allowed us to use it to define a complex simulation like protein folding.”